Booking options
£699 - £9,996

£699 - £9,996
On-Demand course
Has the Computer System Validation Engineer left and you’ve been handed their responsibilities? Do the thoughts of your next audit fill you with dread? CSV can be frustrating but this program will show you how to manage electronic data in a regulated manufacturing/laboratory/clinical environment using the GAMP framework and ensure compliance with FDA’s 21 CFR Part 11, EU Annex 11 or other regulatory guidelines.
Learn the process used to test, validate and formally document that a regulated GxP computerized system application does exactly what it is designed to do.
Go rapidly from beginner to advanced level.
Extend you or your team’s role into CSV projects. Charge higher hourly rates.
Application Deadline: Wednesday 19th July
Study Online part-time
12hrs/week for 10 weeks or choose a faster/slower schedule
Dedicated course leader who’ll check your progress at the end of every week and follow up with you to help you finish the course
Join 3010 learners
Apply NOW. Extend your role to Computer System Validation projects
End-of-week progress checks and follow-up by us to MAKE SURE you or your team finish the course.
Our learners work for the world’s biggest pharma and medical device companies












This program was developed on-site by a team of senior validation engineers, chemical engineers and automation engineers working within an engineering consultancy to train its own engineers and technicians.
Your report will help management make critical decisions about the electronic data generated and managed by the system, (e.g. will it meet the findings from a previous FDA/EMA audit, or to prepare for an upcoming audit, etc). And it will demonstrate:
Your understanding of how the electronic data from the system is managed across the entire life cycle to make sure it is transparent, robust and tamperproof, that it is stored so that it stands the test of time.
That the systems and procedures have been put in place to ensure that nothing crashes and no data is lost and that the necessary electronic records and electronic signatures (ERES) considerations have been included.
You can choose a project from work for this! You will outline the following:
Identify and describe typical business and GMP functions
Schematically represent typical architectural components
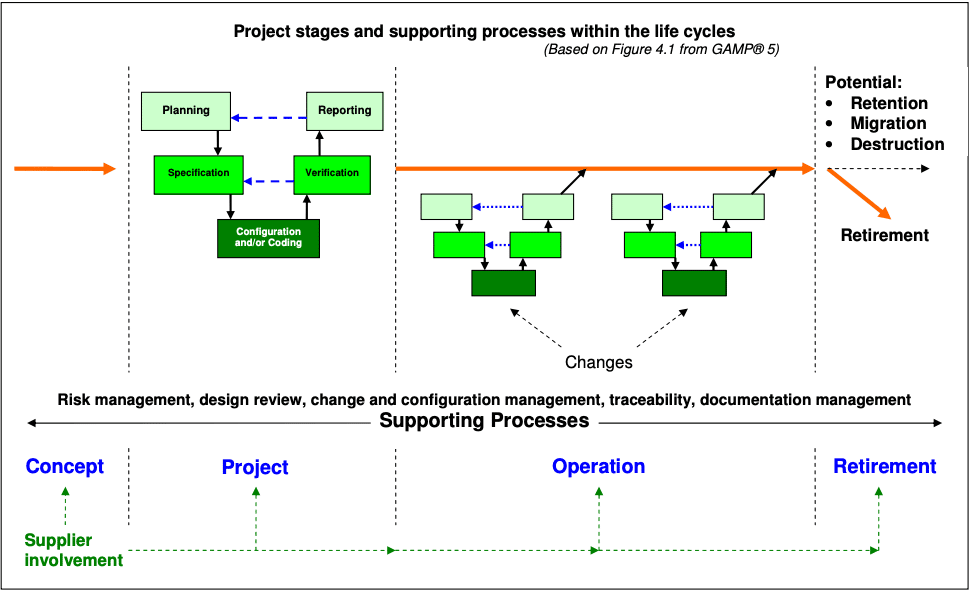
Describe a general approach to achieving compliance and fitness for intended use over the entire lifecycle from:ConceptProjectOperationRetirement
You work with regulated data and have to ensure the following GxP computerized system applications are in compliance with regulatory guidelines such as 21 CFR Part 11 or EU Annex 11.
Laboratory Information Management System (LIMS)Clinical Trial Monitoring SystemsPLC for Controlled Packaging EquipmentSupervisory Control and Data Acquisition (SCADA)Distributed Control System (DCS)Chromatography Data System (CDS)Enterprise Resource Planning (ERP) SystemsManufacturing Execution System (MES)Batch Record SystemBuilding Management Systems (BMS)Cloud base software servicesSpreadsheets
You work in validation, automation or instrumentation and want to extend you or your team’s role, or your consultancy’s offer into Computer System Validation projects.
You want to turn your years of practical work experience into a Certification.
13-year track record. We’ve been delivering this program ONLINE for over 13 years.
One of the single biggest misconceptions of working in Computer System Validation is that you need to be able to code or have a computer science background or be able to program a PLC.
This is not the case as CSV is about managing data accuracy, reliability and integrity, not programming.
However, you do need solid experience using the GxP computer process you will be validating and understand its functional layout and user interface.
The key FDA and international regulations (EudraLex Volume 4 — Annex 11, ICH, PICs) and guidance regarding CSV and which apply to your company
How the CSV process fits into your Software Life Cycle and the purpose of each validation deliverable
The principles of your software quality assurance and how to manage the auditors’ expectations around its key components
The software validation life cycle from design, through construction, installation and live start-up
The regulations governing the use of electronic records and signatures in a regulated environment
The methodology and implementation model for a risk-based approach to CSV
Determine the end-user supplier activities during the lifecycle of a computerized system
Identify where you would use risk-based decision-making throughout the lifecycle of a computerized system
Prepare a configuration management process flow diagram and identify where to use change control
Define the content of typical logs and accompanying records for both incident management and corrective and preventive action (CAPA)
Complete 8-question booklets (one for every week’s worth of content) which will summarise what you have learnt for the week and ensure you retain and understand the information.
Write a 6,000-word technical report to present to senior management on a GxP computerized system application of your choice.
Be able to apply the FDA and international regulations to your current projects
Use Risk-based decision when making an assessment to see what software does or does not require validation, ensure critical risks are identified and the correct level of validation is carried out.
Create key validation deliverables, including validation plans, requirements specifications, test plans, validation tests (IQ, OQ, PQ), trace matrices, test summaries, and validation reports
However, you must have solid experience using at least one GxP computer process and understand its functional layout.
In addition…
This program is highly specialized so you MUST have a solid understanding of Good Manufacturing Practices (GMPs) rules, regulations and guidelines.
AND you are any of the following…
Validation Engineer/Specialist or Senior Validation Manager
PhD, Masters or Degree in Biochemistry, Microbiology, Molecular Biology, Lab/Science, etc
Automation, Control/Instrumentation, Process, Chemical, Manufacturing, etc Engineer or Technician
Quality Assurance or Quality Control Technician or Specialist
Metrology, Maintenance Engineer, Technician or Specialist
Front/Backend Developer
Not sure if you meet the minimum requirements? Contact us.
NOTE: If you don’t have a solid understanding of GMP rules, regulations and guidelines, you need to start with our 6-Week GMP Training For Beginners in the Pharmaceutical Industry. (There’s a special price reduction for this GMP program if taken together with our CSV course. Contact us for details)
CSV is a process used to ensure (and document) that all computer-based systems will produce information or data that meet predefined requirements. If a system meets these requirements, it can be assumed that it is consistently performing in the way it was intended.
The process is used to replace paper with electronic data within highly regulated environments that directly impact public health and safety such as pharmaceutical and medical device manufacturing and make sure the system is:
completely transparent, robust and tamper-proof
and can store those electronic data records so that they stand the test of time
CSV specialists plan, write, implement and review the Computer Systems Validation protocols in place within highly regulated manufacturing industries. Their work is essential to make sure that all computer-based systems are operating as intended (with documents to prove it) to meet regulatory requirements.
There is currently a high demand for trained Computer System Validation Engineers. As a result, salaries are highly competitive.
Need More Detailed Information?
What is Computer System Validation (CSV)?
What is a CSV Engineer?
What are GxP Software Systems? View 10 Examples
Week 1 – Software Categories, Life Cycle Phases, and Operational Activities
1.1 Drivers for Good Automated Manufacturing Practices
In this lesson, we learn about the primary drivers behind the issuing of the Good Automated Manufacturing Practices guidance document and its focus.
1.2 Life Cycle Phases of Computerized Systems
In this lesson, we explore the lifecycle of a computerized system from its early specification to its retirement and upgrade.
1.3 Computerized Systems in Regulated GxP Environments
The objective of this lesson is to be able to describe a computerized system to a regulatory inspector to demonstrate the system is in control and fit for use.
1.4 Good Automated Manufacturing Practices Software Categories
In this lesson, we look at ways to categorize computerized system from infrastructural and off-the-shelf software to configurable and bespoke applications.
1.5 Operational Activities
In this lesson, we take a look at how to maintain a critical computerized system in a state of operational control using standard procedures.
1.6 Handover
This lesson looks at ways to ensure that the environment into which the computerized system is to be received is prepared, to make sure that the system can be used and supported in a controlled manner.
1.7 Product and Process Understanding
This lesson makes recommendations about the application of subject matter expertise when supporting lifecycle activities for a computerized system amongst the end-user and the supplier.
1.8 End-User Activities
This lesson explains the responsibilities of the end-user when it comes to supporting the lifecycle activities for a computerized system.
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 2 – Record Anatomy and Data Flow Analysis
2.1 Record Anatomy
In this lesson, we will describe the structure of a record in terms of the arrangement of data elements, and we will also discuss the ‘electronic record lifecycle.
2.2 Records and Signatures required by 21 CFR Part 211
2.3 PLC Controlled Packaging Equipment
2.4 SupervisoryControlandDataAcquisition(SCADA)
2.5 DataFlowAnalysis
In this lesson, we look at data flow analysis across systems to reveal where integrity could be compromised as the record lives in various computing environments associated with its creation, use, and retention.
2.6 Example Records and Signatures Required by ICH Q7
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 3 – Science Based Quality Risk Management and Validation Planning
3.1 Supplier Activities
This lesson explains the responsibilities of the supplier when it comes to supporting the lifecycle activities for a computerized system.
3.2 Validation Planning
In this lesson, we look at corporate and site level policy documents that define a regulated company’s overall approach to computerized system quality and compliance.
3.3 Science-Based Quality Risk Management
In this lesson, we look at suggestions about where to apply risk management throughout the lifecycle of a computerized system and how to manage the process for various categories of systems.
3.4 Risk Management Considerations – Generic Hazards
In this lesson, we look at an approach to conducting risk assessments on computerized systems based on their impact on product quality, patient safety and data integrity. We also list generic hazards for a computerized system associated with physical/environmental conditions, hardware and software, and human-related.
3.5 Requirements Traceability Matrix (RTM)
In this lesson, we introduce the Requirements Traceability Matrix (RTM); an important project document for tracing all user requirements to design specifications and appropriate verification tests.
3.6 Efficiency Improvements (Continuous Improvements)
In this lesson, we look at suggestions on ways to improve efficiencies throughout the lifecycle of a computerized system.
3.7 Categorization of Laboratory Computerized Systems
This lesson explains the GAMP® system for categorizing Laboratory Computerized Systems.
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 4 – Identify Regulated Records and Signatures, and Impact Assessment of Electronic Records
4.1 HPLC Systems
4.2 Chromatography Data Systems (CDS)
4.3 GxP Records and Signatures Required by 11 CFR Part 820
4.4 Prerequisites for GERM
4.5 Laboratory Information Management System (LIMS)
4.6 Identify Regulated Records and Signatures
In this lesson, we describe why only those records required to meet GxP regulations, or submitted to regulators, should be identified as regulated electronic records.
4.7 Electronic Production Records (EPR)
4.8 Impact Assessment of Electronic Records
This lesson classifies regulated electronic records as High, Medium, or Low based on an assessment of the potential impact of the record on patient safety or product quality.
4.9 Spreadsheets
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 5 – Specification and Verification, Scalable Validation Deliverables and Configuration Management
5.1 Organizational Change – Impact During Project Lifecycle
This lesson looks at various ways to manage projects during episodes of major organizational changes.
5.2 Outsourced IS/IT Environment
Here we look at the contract considerations when we choose to outsource control of our IS/IT systems to a third-party vendor.
5.3 I T Compliance – Key Concepts, and Infrastructure Elements
In this lesson, we look at ways to standardize our IT Infrastructure Elements to facilitate and somewhat ‘modularize’ our compliance strategy.
5.4 Development versus Implementation Life Cycle
In this lesson, we describe how to implement various computerized systems based on their software category; where from a user’s perspective the lower categories require less vendor input and where the higher categories require more.
5.5 Specification, Design and Verification
In this lesson, we review the ASTM International standard E 2500 – 07 ‘Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment’.
5.6 Testing Documentation Structure & Verification Terminology
In this lesson, we look at good practices regarding the organization of testing documentation on a project and we also examine legacy and modern terms for testing documentation used in the medicinal products’ industries.
5.7 Scalable Validation Deliverables
This lesson recommends a range of scaleable validation deliverables over the range of software categories where we see increasing intricacy with increasing category designation.
5.8 Patch and Update Management
In this lesson, we consider suitable management strategies for the implementation of software patches and upgrades to an existing computerized system operating in a regulated environment.
5.9 Operational Change and Configuration Management
This lesson describes how to manage the configuration of a computerized system in a regulated environment during the operation and maintenance phase and keep it current and relevant where we’re in a period of continuous change and upgrading.
5.10 Repair Activity
In this lesson, we discuss a process by which non-functional systems are returned to a functional state under the control of a repair activity procedure.
5.11 Periodic Review
In this lesson we’re going to look at a method to ensure a computerized system remains compliant with regulatory requirements throughout its operational life, remains fit for intended use, and continually satisfies company policies and procedures.
5.12 Backup and Restore
In this lesson, we describe a mechanism to protect electronic information assets against loss of original data and subsequent accurate restoration of assets when required.
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 6 – Good Electronic Records Management Transactions, and Audit Trails
6.1 Good Electronic Records Management Transactions
In this lesson, we look at current good practices associated with good electronic records management including transactions, audit trails, sequence checks, electronic signatures, and continuous session system access.
6.2 Audit Trails
In this lesson, we explore how the application of audit trails improve information quality and reduce information loss from activities such as overwriting of data attributes.
6.3 AutoCAD Used for Managing Pack Drawings
6.4 Building Management Systems (BMS)
6.5 21 CFR Part 211 – Subparts D and J
In this lesson we look at the FDA’s predicate GMP rule 21 CFR Part 211 specifically Subparts D and J. Subpart D relates to ‘Equipment’ and how subpart J relates to ‘Records and Reports’.
6.6 FDA 21 CFR Part 11 – ‘Electronic Records; Electronic Signatures (ERES)’
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 7 – Electronic Data Archiving, Business Continuity Management, and System backup, Archival, and Disaster Recovery
7.1 Electronic Data Archiving
In this lesson, we describe a suitable data archiving strategy for moving data that is no longer actively used in the active environment where it was created to a separate data storage area for long-term retention.
7.2 Typical Tasks Supporting Validation
Here we look at recommendations from the FDA regarding tasks that may support the validation of a computerized system in a regulated environment.
7.3 Security Management
In this lesson, we define the controls required for securing a computerized system in an operational environment.
7.4 Business Continuity Management
In this lesson, we discuss how to regain access to an IT system and its data following a disaster, and how to restore critical business processes following a disruption while continuing to provide products or services.
7.5 Relationship Between System Backup, Archival, and Disaster Recovery
In this lesson we look at the inter-relationship amongst the accurate and reproducible backing up of digital assets (data and software), the archiving of closed business transactions and master data relocation to an external system, and the regaining of access to an IT system (including software, hardware and data) following a disaster.
7.6 System Retirement, Decommissioning and Disposal
This lesson describes appropriate controls that need to be in place when removing a computerized system from day to day use through obsolescence or replacement.
7.7 Copies of Records
This lesson considers methods on how to preserve the content and meaning of an electronic record when making a copy.
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 8 – Controls to Maintain Electronic Record Integrity, and Risk Controls for Electronic Signatures
8.1 Complying with 21 CFR Part 11 ERES – Types of Controls Required
8.2 ERES – Key Areas for Guidance – Where to Apply Electronic Signatures
This video explores examples of where to apply electronic signatures required by predicate rules and required by internal procedures.
8.3 Batch Record Systems
8.4 Enterprise Resource Planning (ERP) Systems
8.5 Controls to Maintain Electronic Record Integrity
In this lesson, we examine controls to maintain electronic record integrity when creating and
storing records, transmitting records, and archiving records, and also controls for signature certification/ authentication and signature link integrity.
8.6 Risk Controls for Electronic Records
8.7 Risk Controls for Electronic Signatures
In this lesson, we look at risk controls for electronic signatures in terms of instruction for the information associated with a signed electronic record, e-signatures security certificates, and signature controls.
8.8 User ERES Responsibilities
In this lesson, we look at the end-user’s responsibilities associated with electronic records and electronic signatures.
8.9 Supplier ERES Responsibilities
In this lesson, we look at the supplier’s/vendor’s responsibilities associated with electronic records and electronic signatures.
Deliverables
Complete a question booklet that will summarise what you have learnt for the week and help ensure you retain and understand the information.
Week 9 & 10 Complete an End of Module Assignment
Complete a 6,000-word technical report written at a Masters/Graduate level, on a GxP system of your choice. You can choose a project from work for this! The assignment should outline the controls that have been put in place on an existing/new/upgraded system, to generate and manage electronic data from the system for the benefit of the patients who use the products manufactured/approved/released by the system.
It should be written for a senior management audience so as to give them the necessary information they need to make informed management decisions about the electronic data generated and managed by the system, (e.g. will it meet the findings from a previous FDA audit, or to prepare for an upcoming one, etc) to support the manufacturing of safe and effective medicines and medical devices for patients.
The structure of the report should include details on the LifeCycle of your chosen data management system, to confirm your understanding and reflect your ability to apply your learning from the CSV programme to a real-world situation:
Concept of the chosen data management system, including what the original URS wanted to achieve in terms of data generation and management, the risk management assessment to support the critical decisions made in the choices that were taken in the design/specification of the data management, the document control that was put in place to record the data generated for future audit.
The project itself has to be defined, using tools like the V-model to describe the steps taken in the project to gather data and how the IQ-OQ-PQ phases ensured that the system installed (or to be installed) is fit for purpose to manage/control the electronic data generated (and how it met or modified the requirements of the URS).
The learnings (or expected learnings) from the Operation of the system have to be described noting key data management areas such as CAPA’s and Maintenance, and the systems used to ensure that nothing crashes and no data is lost.
The preparations for the Retirement of the system at some time in the future have to be described, noting how the electronic data can be migrated to another system (or deleted).
The assignment must demonstrate:
Your understanding of how the electronic data from the system is managed to make sure it is transparent, robust and tamperproof, that it is stored so that it stands the test of time,
That the systems and procedures have been put in place to ensure that nothing crashes and no data is lost, and that the necessary ERES considerations have been included.

Full-Time Validation Lead
Lecturer, Technological University Dublin, Ireland
Senior Associate, GetReskilled
Dr. Joe Brady is a full-time practicing Validation Lead and an assistant lecturer with Technological University Dublin (TU Dublin), in the School of Chemical and Pharmaceutical Sciences. Joe is a certified trainer and highly experienced in competency-based training. He designs and prepares educational modules and full academic courses ranging from MSc, MEngSc. BSc, to Certificate level, for a range of academic institution.
He is also a supervisor for MSc/MEngSc and PhD theses. Joe has over twenty years of project experience in the pharmaceutical, biopharmaceutical and medical device industries in Ireland, Singapore, China, The Netherlands, France and the USA.


“All sections of the course were explained well and the online delivery allowed for a flexible study plan which was very beneficial when working alongside study. The end of module assignments targeted key points in the lectures which was useful when writing the final assignment. I found this course very useful in my current role and would highly recommend it to anyone looking to gain knowledge in Computer System Validation.”


“I have found the Computer System Validation course to be both interesting and helpful in my current role. The individual sections of the course are easy to follow. The delivery of the course is really straight forward. During the course there has been times when I have been busy with work and have not been able to study. The structure of the course allows me to be flexible in my study.”


“I would highly recommend anyone wishing to gain an education in the Computer System Validation sector to do this course. For me it has been very useful studying on-line in order to do the course in my own time. Lessons are very clear and the assignment deadlines make you disciplined to meet the deadlines set.”

With every GetReskilled ONLINE program;
We use one centralized platform (Moodle) where you can log into your classroom anytime. Each week, you’ll watch videos and complete a series of quizzes, tests, interactive activities, and projects. The course materials are available 24/7 and nothing requires you to be online at a specific day or time. i.e there are NO ZOOM classes and NO WEBINARS! Study anywhere, anytime, for example after the kids have gone to bed or on the weekend.
Your working schedules are unpredictable so we offer flexible delivery. Slow down, speed up or pause the delivery of the program.
We release only one week’s worth of material at a time and then MANUALLY check your activity logs at the end of every week to make sure that you are keeping up with your work.
You’ll have a dedicated course leader who will email or telephone you if it looks like you’re starting to fall behind. They will work with you to develop a study plan to get you back on schedule and finish the course.
This all helps us to spot any potential issues early and helps you completely finish the program.
Start your application
Get certified and extend you or your team’s role to CSV Projects
Apply NOW. Extend your role to Computer System Validation projects
End-of-week progress checks and follow-up by us to MAKE SURE you or your team finish the course.
USA/World
Call: +1 (617) 901 9268
Ireland
Call Geraldine: +353 (0)21 2409016
Earn by successfully completing a 6,000-word technical report on a GxP computerized system application of your choice and get a Certificate of Award in Commissioning & Qualification (IQ OQ PQ) of Equipment and Systems.
Add details of your certificate to your CV/Resume or your LinkedIn profile.


GetReskilled is an awarding-winning education company. We'll retrain or upskill you ONLINE for a higher-paying career or a promotion in the Pharmaceutical and Medical Devi...