- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
10873 Professional Development courses in Goole delivered On Demand
Emotional Intelligence, Organisational Behaviour, REBT, Psychology & Career Development - 20 Courses Bundle
By NextGen Learning
Get ready for an exceptional online learning experience with the Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development bundle! This carefully curated collection of 20 premium courses is designed to cater to a variety of interests and disciplines. Dive into a sea of knowledge and skills, tailoring your learning journey to suit your unique aspirations. The Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development is a dynamic package, blending the expertise of industry professionals with the flexibility of digital learning. This Emotional Intelligence package offers the perfect balance of foundational understanding and advanced insights. Whether you're looking to break into a new field or deepen your existing knowledge, the Emotional Intelligence package has something for everyone. As part of the Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development package, you will receive complimentary PDF certificates for all courses in this bundle at no extra cost. Equip yourself with the Emotional Intelligence bundle to confidently navigate your career path or personal development journey. Enrol today and start your career growth! This Emotional Intelligence Bundle Comprises the Following CPD Accredited Courses: Emotional Intelligence Organizational Behaviour, HR and Leadership Level 3 Coaching & Mentoring Course Master Emotional Intelligence for Crucial Moments Rational Emotive Behaviour Therapy (REBT) Certificate Influencing and Negotiating Personal and Networking Skills Anger Management and Conflict Resolution Program Career Development and Passion Positive Psychology Masterclass Goal Setting, Motivation, and Resilience for Life Using Mindfulness at Work for Productivity Self-Help Psychology: Mental Freedom Self Confidence & Self Esteem Workplace Productivity Training Career Development Plan Fundamentals CV Writing and Job Searching Learn to Level Up Your Leadership Networking Skills for Personal Success Ace Your Presentations: Public Speaking Masterclass Learning Outcome: Gain comprehensive insights into multiple fields. Foster critical thinking and problem-solving skills across various disciplines. Understand industry trends and best practices through the Emotional Intelligence Bundle. Develop practical skills applicable to real-world situations. Enhance personal and professional growth with Emotional Intelligence. Build a strong knowledge base in your chosen course via Emotional Intelligence. Benefit from the flexibility and convenience of online learning. With the Emotional Intelligence package, validate your learning with a CPD certificate. Each course from Emotional Intelligence bundle holds a prestigious CPD accreditation, symbolising exceptional quality. The materials, brimming with knowledge, are regularly updated, ensuring their relevance. This Emotional Intelligence bundle promises not just education but an evolving learning experience. Engage with this extraordinary collection, and prepare to enrich your personal and professional development. Embrace the future of learning with Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development, a rich anthology of 15 diverse courses. Each course in the Emotional Intelligence bundle is handpicked by our experts to ensure a wide spectrum of learning opportunities. This Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development bundle will take you on a unique and enriching educational journey. The Emotional Intelligence bundle encapsulates our mission to provide quality, accessible education for all. Whether you are just starting your career, looking to switch industries, or hoping to enhance your professional skill set, the Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development bundle offers you the flexibility and convenience to learn at your own pace. Make the Emotional Intelligence package your trusted companion in your lifelong learning journey. CPD 200 CPD hours / points Accredited by CPD Quality Standards Who is this course for? The Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development bundle is perfect for: Lifelong learners looking to expand their knowledge and skills. Professionals seeking to enhance their career with CPD certification. Individuals wanting to explore new fields and disciplines. Anyone who values flexible, self-paced learning from the comfort of home. Career path Unleash your potential with the Emotional Intelligence, Organizational Behaviour, REBT, Psychology & Career Development bundle. Acquire versatile skills across multiple fields, foster problem-solving abilities, stay ahead of industry trends. Ideal for those seeking career advancement, a new professional path, or personal growth. Embrace the journey with the Emotional Intelligence bundle package. Certificates Certificate Of Completion Digital certificate - Included Certificate Of Completion Hard copy certificate - Included You will get a complimentary Hard Copy Certificate.

Levers of Project Agility: Effective Sponsorship
By IIL Europe Ltd
Levers of Project Agility: Effective Sponsorship Levers of Project Agility: Effective Sponsorship You may be using agile processes in your projects, even have extended the use of agile management practices into other areas of business. However, lack of purposeful and appropriate sponsorship can stifle most projects. An under-engaged or over-enthusiastic sponsor can demotivate the team, slowdown decision making and disrupt even best agile processes. In this talk, we will examine the crucial role of the sponsor, their desired attributes and their relation with the project manager, product owner and scrum master to identify the risk factors and provide tips and tools for avoiding pitfalls and having effective sponsors. You may be agile, but a poor sponsor can still hamper project success. We'll examine this crucial role, ideal attributes, and provide tips to maximize sponsor effectiveness. This and other IIL Learning in Minutes presentations qualify for PDUs. Some titles, such as Agile-related topics may qualify for other continuing education credits such as SEUs, or CEUs. Each professional development activity yields one PDU for one hour spent engaged in the activity. Some limitations apply and can be found in the Ways to Earn PDUs section that discusses PDU activities and associated policies. Fractions of PDUs may also be reported. The smallest increment of a PDU that can be reported is 0.25. This means that if you spent 15 minutes participating in a qualifying PDU activity, you may report 0.25 PDU. If you spend 30 minutes in a qualifying PDU activity, you may report 0.50 PDU.

Whether you're drawn to supporting others through difficult times or curious about the inner workings of the human mind, this CPD-accredited Counselling Diploma – 3 Courses Bundle offers a solid introduction to modern counselling techniques. With three distinct yet interlinked courses — Counselling Diploma, Life Coaching and Counselling, and Online CBT (Cognitive Behavioural Therapy) — this bundle presents a well-rounded insight into emotional support, mindset management, and behavioural change theory. You’ll explore core counselling approaches, structured life coaching concepts, and CBT methodologies — all delivered in an accessible, online-friendly format. The course is ideal for those seeking a foundational understanding of mental wellbeing strategies, whether for personal development or to enrich existing knowledge. No textbooks to lug around, no overly philosophical waffle — just straight-talking, professionally structured modules built to help you understand the what, why, and how of supporting mental health. If your idea of progress includes helping people untangle their thoughts without having to leave your sofa, this could be the beginning of something meaningful. These comprehensive courses are available in this Counselling Diploma - CPD Accredited 3 Courses Bundle Course 01: Counselling Diploma Course 02: Life Coaching and Counselling Course 03: Online CBT (Cognitive Behavioural Therapy) Course **Special Offer: Free PDF and Hard Copy Certificates** Key Benefits Get instant PDF and Hard Copy certificate Fully online courses Developed by qualified professionals Self-paced learning and laptop, tablet, and smartphone-friendly 24/7 Learning Assistance Course Curriculum: Counselling Diploma Module 01 : Counselling and Counsellors Module 02 : Theories and Models of Counselling (Part 1) Module 03 : Theories and Models of Counselling (Part 2) Module 04 : Legal, Cultural and Ethical Issues in Professional Counselling Module 05 : Forms of Psychotherapy Module 06 : Engaging and Assessing the Patients Module 07 : Helping the Client in Crisis Module 08 : Crisis Intervention Module 09 : Low Intensity CBT Treatment Module 10 : Medication and Therapy in the Treatment of Mental Illness Module 11 : Communication Skills and Empathy in Counselling Course Assessment You will immediately be given access to a specifically crafted MCQ test upon completing each Counselling Diploma - CPD Accredited 3 Courses Bundle bundle course. For each test, the pass mark will be set to 60%. Accredited Certificate After successfully completing this Counselling Diploma - CPD Accredited 3 Courses Bundle course, you will qualify for the CPD Certification Service (CPD UK) certified certificate from Training Express. Disclaimer: The CPD approved course is owned by E-Learning Solutions Ltd and is distributed under license. Why Choose The CPD Certification Service Accredited Course? With 25 years of experience in the Continuous Professional Development sector, CPD Certification Service (CPD UK) is the leading CPD accreditation organisation in the UK, working across all industry sectors. The CPD Certification Service provides recognised independent CPD accreditation compatible with global CPD requirements. Hundreds and thousands of professionals recognise the CPD Certified symbol as the qualitative benchmark that reflects and sets the industry standards. CPD UK evaluates learning activities to the highest standards and courses are certified against the universally accepted structured checklist. CPD 30 CPD hours / points Accredited by The CPD Certification Service Who is this course for? The Counselling Diploma - CPD Accredited 3 Courses Bundle training is ideal for highly motivated individuals or teams who want to enhance their skills and efficiently skilled employees. Requirements There are no formal entry requirements for the course, with enrollment open to anyone! Career path Learn the essential skills and knowledge you need to excel in your professional life with the help & guidance from our Counselling Diploma - CPD Accredited 3 Courses Bundle training.

Lab Technician: Lab Technician Course Unlock the Power of Lab Technician: Lab Technician Course: Enrol Now! Do you want to develop your skills as a lab technician or pursue a career in it? If yes, sign up for this Lab Technician: Lab Technician Course to get the knowledge and abilities required to become more organised and productive. The goal of the Lab Technician: Lab Technician Course was to provide participants a thorough grasp of Lab technicians with a focus on legal ideas, best practices, and underlying competence. The lab technician course explains the function of the Lab Technician: Lab Technician Course as well as laboratory safety, ethics, equipment, and instruments. As a lab technician, you will be familiar with medical language, paperwork, and specimen collecting and management. This Lab Technician: Lab Technician Course explains all the responsibilities of a lab technician in pathology, professional growth job choices, and much more. Sign up for the Lab Technician: Lab Technician Course to increase your output at work. Main Course: Lab Technician Course Free Courses included with Lab Technician: Lab Technician Course Along with Level 3 Certificate in Nutrition Course you will get free Medical Terminology Course Along with Level 3 Certificate in Nutrition Course you will get free Medical secretary & receptionist Course Special Offers of this Lab Technician: Lab Technician Course: This Lab Technician: Lab Technician Course includes a FREE PDF Certificate. Lifetime access to this Lab Technician: Lab Technician Course Instant access to this Lab Technician: Lab Technician Course 24/7 Support Available to this Lab Technician: Lab Technician Course Lab Technician: Lab Technician Course With an emphasis on Lab Technician: Lab Technician Course legal concepts, Lab technician best practices and the underlying expertise, the Lab Technician: Lab Technician Course aimed to provide participants with an in-depth understanding of Lab technicians. The Lab Technician: Lab Technician Course role, laboratory safety, ethics, equipment and instruments are explained in the Lab Technician: Lab Technician Course. You will know the medical terminology, documentation, specimen collection and handling as a Lab technician. All the roles of a Lab Technician in pathology, professional development career paths and many more are explained in this lab Technician. Who is this course for? Lab Technician: Lab Technician Course Enrolling in this Lab Technician: Lab Technician Course will enhance your resume and provide you with the chance to research the following positions Requirements Lab Technician: Lab Technician Course To enrol in this Lab Technician: Lab Technician Course, students must fulfil the following requirements: Good Command over English language is mandatory to enrol in our Lab Technician: Lab Technician Course. Be energetic and self-motivated to complete our Lab Technician: Lab Technician Course. Basic computer Skill is required to complete our Lab Technician: Lab Technician Course. If you want to enrol in our Lab Technician: Lab Technician Course, you must be at least 15 years old. Career path Lab Technician: Lab Technician Course This training curriculum will be helpful to UK residents looking for new Lab Technician: Lab Technician Course work prospects.

SUB06: Variations to Marketing Authorisations in Europe
By Zenosis
Changes to the terms of marketing authorisations for medicinal products, called variations in Europe, must be notified to or approved by the relevant regulatory authorities. Variations include changes to the composition of products, their manufacturing processes, the way they are used, or the indications for which they are authorised. Common approaches are adopted within the European Economic Area to variations to marketing authorisations approved through the Centralised, Decentralised or Mutual Recognition Procedures. Recent legislation has substantially modified the regulatory requirements and extended them to purely national authorisations by member states. This module, which is fully up to date with the new legislation, covers the classification of variations into their several types and the regulatory requirements, guidance and procedures to be followed for each type.
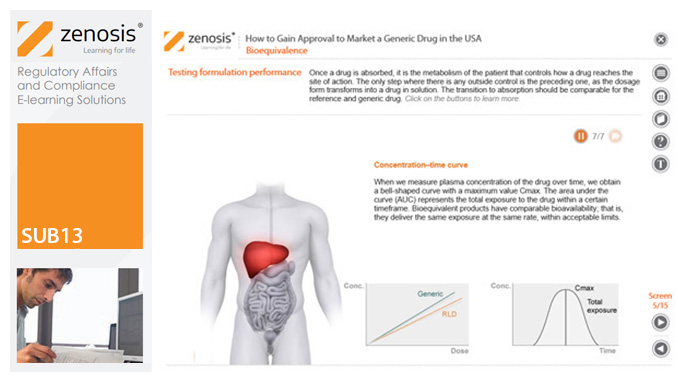
SUB13: How to Gain Approval to Market a Generic Drug in the USA
By Zenosis
This module outlines the legislative and regulatory context for the development of generic drugs and describes the essential role of the Abbreviated New Drug Application (ANDA) in gaining marketing approval. The use of information in the ‘Orange Book’ is explained, as is the role of patent certification in the application. The importance of establishing bioequivalence between a generic and its reference product is emphasised. The module specifies the content and format requirements for an ANDA submission and describes the FDA’s review and approval process. An outline is given of the Generic Drug User Fee Amendments (GDUFA) and the law’s effects on industry players.
SUB14: The Regulatory Pathway to Licensure of Follow-on Biologics (Biosimilars) in the USA
By Zenosis
The regulation of biological medicinal products is governed by different laws from those that apply to small-molecule synthetic drugs. Producing faithful copies of therapeutic proteins is more challenging than producing generic drugs. The US legal framework for the licensure of follow-on biologics, and accompanying regulatory guidance from the Food and Drug Administration (FDA), have been established only in recent years.

SUB15: The Biologics License Application (BLA) for Marketing Approval in the USA
By Zenosis
This module describes the requirements that must be met to obtain licensure of a biological product. Subjects covered include the regulatory context, the content and format of the BLA submission, the review process, and provisions for expedited development and review.

VAL06: Computer Systems Validation, Part 1: Planning
By Zenosis
In the medicines and healthcare products industries, computerised systems used in automated manufacturing or laboratory processes to which Good Manufacturing Practice requirements apply need to be validated. This module describes the planning of such validation. It follows the work of a pharmaceutical company's team as they validate the dispensary control system for a new production line.

VAL05: Equipment Cleaning Validation
By Zenosis
Manufacturers of medicines and healthcare products must establish, validate and maintain an equipment cleaning programme. This is a regulatory requirement because validated cleaning procedures contribute to the assurance of product purity and safety. This module provides a comprehensive account of equipment cleaning validation requirements and procedures. It follows the work of a pharmaceutical company's validation team as they establish and validate the cleaning program for a new production line.