- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
281 Pharmacology courses delivered On Demand
CT03e - Clinical trial investigator’s GCP responsibilities
By Zenosis
A clinical investigator is responsible for conducting the clinical trial in compliance with the study protocol, GCP, medical ethics, and applicable legal requirements. The clinical research community expects that investigators and clinical staff are fully trained in GCP. Duties and functions discussed in this short course include: provision of adequate resources; liaison with IRB/IEC; compliance with protocol; management of investigational product(s), informed consent and data records; and safety reporting.
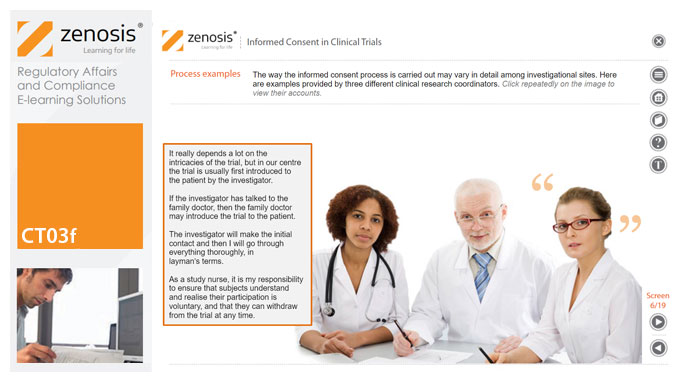
CT03f - Informed consent in clinical trials
By Zenosis
Informed consent in clinical research is an ethical and regulatory requirement. A research subject must enter a study voluntarily, be informed about risks and benefits, and understand the difference between investigation and treatment. Subjects must not be coerced into enrolment, nor must they be enticed by exaggerated claims of benefit. Before they can enrol, all potential subjects must agree, in writing, to participate. In addition to ethical and regulatory imperatives, the potential for litigation by subjects further highlights the importance of rigorous adherence to informed consent principles. In this short course we set out the principles and requirements and provide examples of practical issues confronting healthcare professionals and subjects.
CT04b - Clinical protocol design
By Zenosis
Clinical trial protocols are an essential part of clinical trial design. Protocol documents are critical to conducting safe and cost-effective investigations. Protocol documents are large and complex, containing comprehensive information relating to purpose, design and conduct of a clinical trial. Aspects of a protocol include patient eligibility criteria, and treatment specifications. This short course provides an overview of clinical trial protocols. Opportunities to improve a clinical trial protocol for regulatory approval are also discussed.
GMP01b - Principles of GMP
By Zenosis
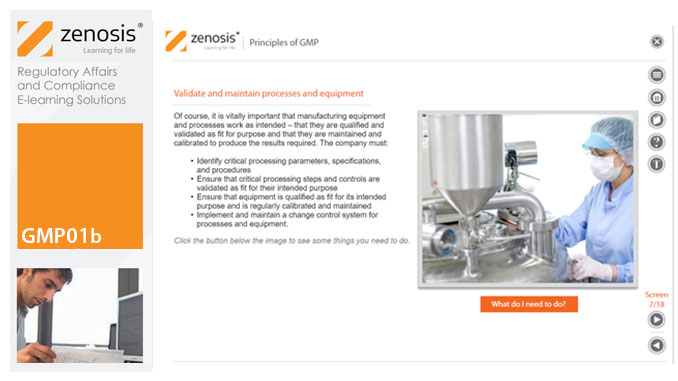
In this short course we present an overview of the main principles of GMP, and we outline some things that manufacturing personnel need to do to comply with requirements. We identify the principal goals of GMP as: prevention of contamination; prevention of mix-ups; scrupulous documentation; validation and maintenance of processes and equipment; quality assurance by an independent unit; and training. We place GMP in the context of a company’s quality management system.
CT04e - Statistical elements of clinical trials
By Zenosis
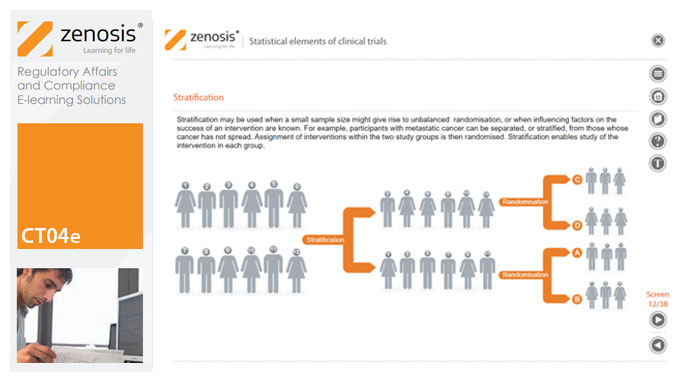
Analytical statistical elements are essential concepts in the design of clinical trials. This analysis helps us to understand whether a conclusion from a study of a sample of the target population applies generally to that population as a whole. In particular, it helps us to answer the question: Did the treatment effect in the given study occur just by chance? The statistical elements of a well-controlled study minimise the chances of drawing the wrong conclusions, by providing clear thresholds for such errors. The basic statistical elements of a clinical trial include eligibility criteria, randomisation, sample size, power, and blinding, and these are discussed in this short course.
CT03c - Clinical trial documentation
By Zenosis
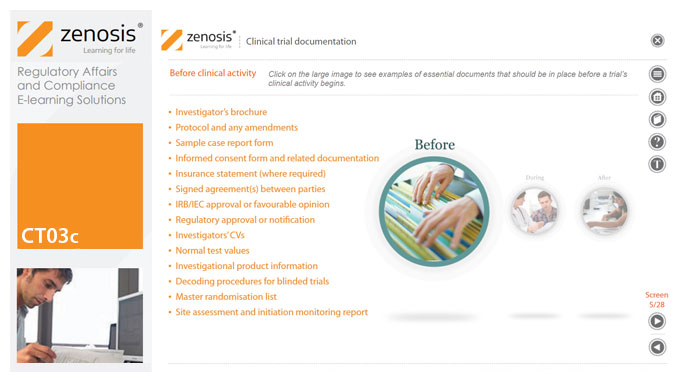
Regulatory authorities tend to abide by the maxim that ‘If it isn’t documented, it didn’t happen’. Rigorous documentation of all aspects of a clinical trial is necessary to provide evidence of GCP and compliance with regulatory requirements, as well as enabling effective management of the trial. In this short course we describe important examples of the documents designated by ICH GCP as essential to the conduct of a clinical trial.
GMP01c - Hygiene, cleaning, and sanitation
By Zenosis
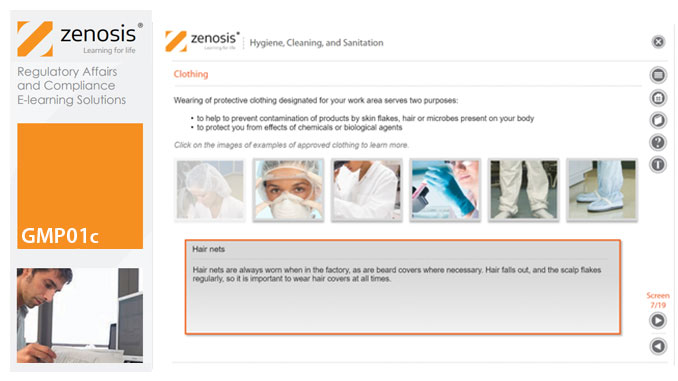
Prevention of contamination is one of the most important goals of GMP. Contamination of product is often difficult to detect, so GMP rules emphasise preventive measures, including: attention to personal health and hygiene, and the wearing of special clothing, by staff; and cleaning and sanitation of premises and equipment. In this short course we set out the basics of GMP requirements in these vital areas.
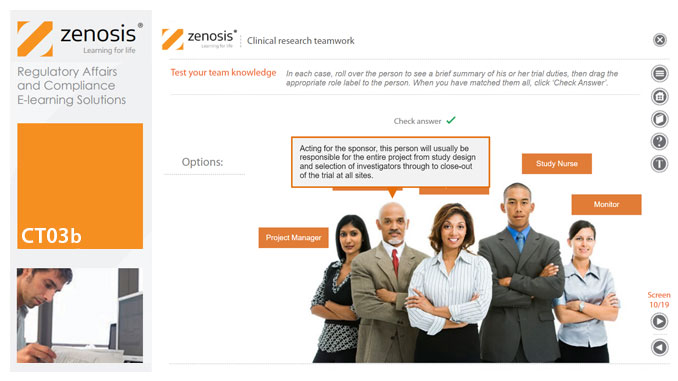
CT03b - Clinical research teamwork
By Zenosis
A clinical trial, particularly a late-phase commercial study, is a major project requiring collaboration between the sponsor and staff or contractor, on the one hand, and the clinical investigator(s) and other healthcare professionals on the other. Good communication among all parties is essential. In this short course we introduce the major roles in a typical clinical research project and outline their duties.
CT03a - ICH, harmonisation, and principles of Good Clinical Practice
By Zenosis
Good Clinical Practice (GCP) is a set of internationally recognised ethical and scientific quality requirements for designing, conducting, recording and reporting clinical trials. Compliance with GCP principles is required by regulatory authorities in many countries for the authorisation of clinical trials and the acceptance of their data in applications for marketing approval. The International Council for Harmonisation's guideline E6, often referred to as ICH GCP, is the international standard specification for Good Clinical Practice. In this short course we describe the ICH’s role in the harmonisation of regulations, introduce its guideline E6, and set out the principles of GCP.
CT04d - Clinical trial endpoints
By Zenosis
In clinical trials, endpoints are measurements to evaluate the results of a new treatment, at an individual patient level. The study data can be extrapolated to patient populations on the basis of clinical similarities to patients participating in the trial. When clinical trial data have been obtained, focus is on the trial endpoints; more specifically, the focus is on whether the trial met or failed the primary endpoint specified before the trial started. The purpose and various types of endpoints are discussed in this short course.