- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
271 Pharmacology courses in Cardiff delivered On Demand
GMP06: Good Manufacturing Practice in Packaging Medicinal Products
By Zenosis
Packaging for medicinal products is subject to Good Manufacturing Practice rules similar to those for the products themselves. In this module we describe the functions that packaging must fulfil and the quality controls that are applied to packaging materials and operations. We set out the requirements for control of printed materials. We describe preparation, in-process control, and completion of a packaging run. Finally, we explain how to carry out reconciliation of packaging materials.

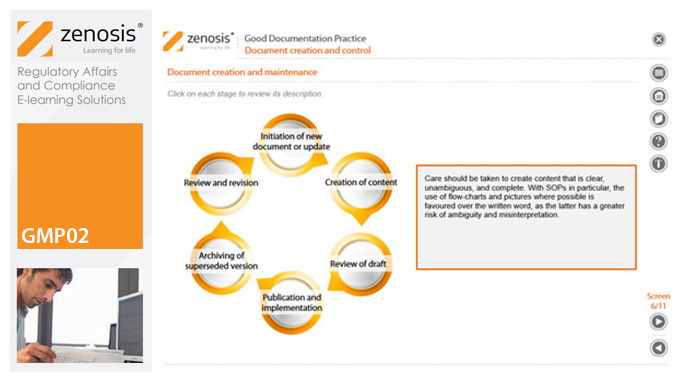
GMP02: Good Documentation Practice
By Zenosis
Good Manufacturing Practice (GMP) for medicinal products relies on documentation. Good Documentation Practice (GDocP) is that part of GMP that applies to the creation, maintenance, use, and retention of documents to provide assurance of the quality of products.
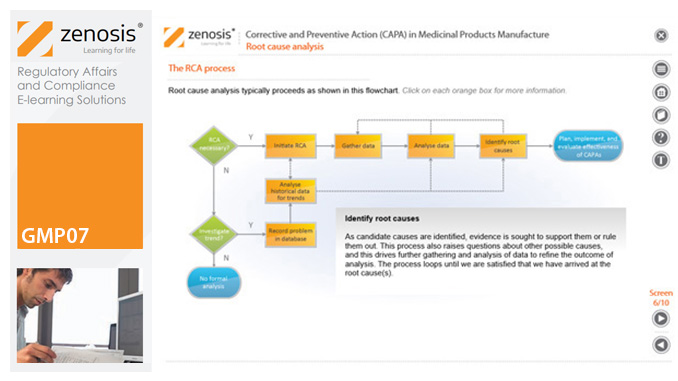
GMP07: Corrective and Preventive Action (CAPA) in Medicinal Products Manufacture
By Zenosis
A company’s Corrective and Preventive Action (CAPA ) system establishes how personnel should deal with manufacturing problems that have occurred or that may occur if not prevented. This module explains the principles of corrective and preventive action and describes typical CAPA procedure. It goes on to introduce root cause analysis and outline the role of progress tracking, escalating, and trending of CAPA procedures.
ESS02: Essentials of Monoclonal Antibodies
By Zenosis
This module will introduce you to monoclonal antibodies, explaining how they work, how they are made, and the many uses to which they are put.

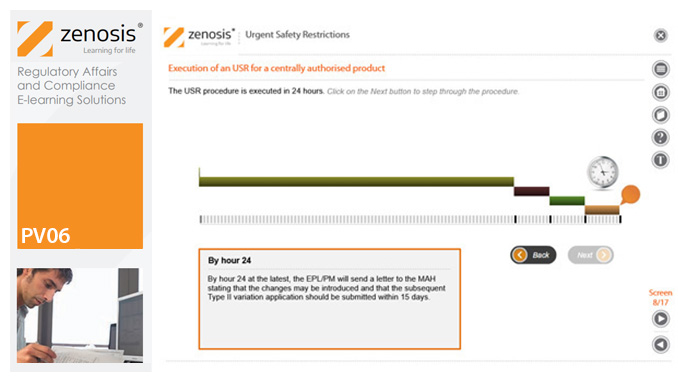
PV06: Urgent Safety Restrictions
By Zenosis
An Urgent Safety Restriction (USR) is a regulatory action taken, in response to a safety signal, to make an interim change to the terms of the marketing authorisation for a medicinal product in Europe. This module describes the principles and procedures for USRs.
GXP01- Good Practices (GxP) in Drug Development and Manufacturing
By Zenosis
This short entry-level module introduces the learner to good practices (GxP) in drug development and manufacturing. It outlines how the industry operates and how it is regulated. It identifies regulatory authorities and other important sources of guidance on Good Manufacturing Practice (GMP), Good Clinical Practice (GCP), and Good Laboratory Practice (GLP).

GXP01: Good Practices (GxP) in Drug Development and Manufacturing
By Zenosis
This short entry-level module introduces the learner to good practices (GxP) in drug development and manufacturing. It outlines how the industry operates and how it is regulated. It identifies regulatory authorities and other important sources of guidance on Good Manufacturing Practice (GMP), Good Clinical Practice (GCP), and Good Laboratory Practice (GLP).

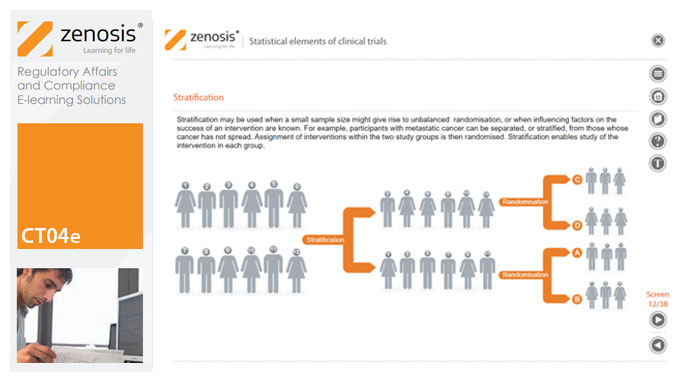
CT04e - Statistical elements of clinical trials
By Zenosis
Analytical statistical elements are essential concepts in the design of clinical trials. This analysis helps us to understand whether a conclusion from a study of a sample of the target population applies generally to that population as a whole. In particular, it helps us to answer the question: Did the treatment effect in the given study occur just by chance? The statistical elements of a well-controlled study minimise the chances of drawing the wrong conclusions, by providing clear thresholds for such errors. The basic statistical elements of a clinical trial include eligibility criteria, randomisation, sample size, power, and blinding, and these are discussed in this short course.
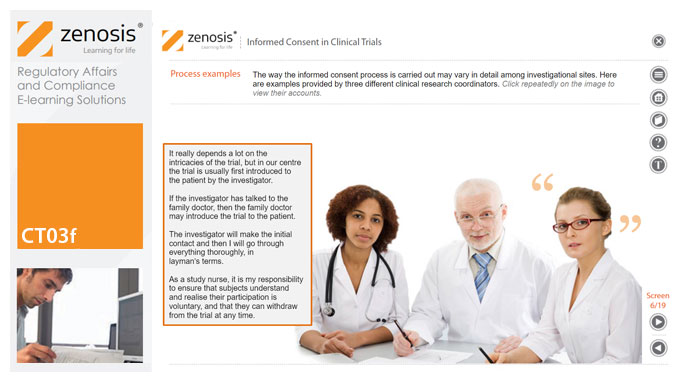
CT03f - Informed consent in clinical trials
By Zenosis
Informed consent in clinical research is an ethical and regulatory requirement. A research subject must enter a study voluntarily, be informed about risks and benefits, and understand the difference between investigation and treatment. Subjects must not be coerced into enrolment, nor must they be enticed by exaggerated claims of benefit. Before they can enrol, all potential subjects must agree, in writing, to participate. In addition to ethical and regulatory imperatives, the potential for litigation by subjects further highlights the importance of rigorous adherence to informed consent principles. In this short course we set out the principles and requirements and provide examples of practical issues confronting healthcare professionals and subjects.
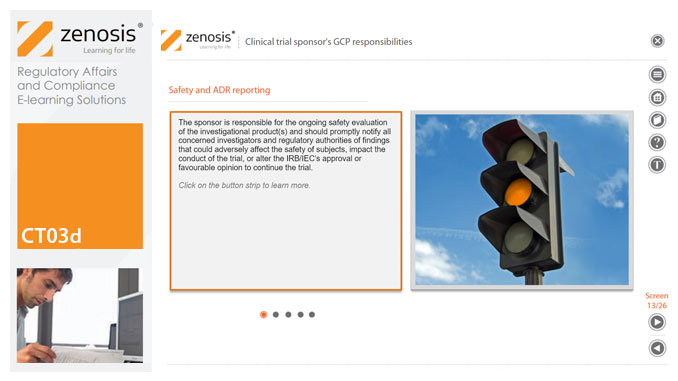
CT03d - Clinical trial sponsor’s GCP responsibilities
By Zenosis
The sponsor of a clinical trial takes responsibility for its initiation, management, and/or financing. A sponsor may transfer any or all of the sponsor’s trial-related duties and functions to a contract research organisation, but the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor. Duties and functions discussed in this short course include trial design, selection of investigators, QA and QC, data handling and record keeping, finance and compensation, regulatory submissions, management of investigational product(s), safety reporting, monitoring, audit, dealing with noncompliance, and clinical trial reports. ICH guideline E6 (revision 2) encourages sponsors to adopt a risk-based approach to managing the quality of trials. We discuss this approach in general, and aspects such as risk-based monitoring in particular.