- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
98 Drug Safety courses delivered Online
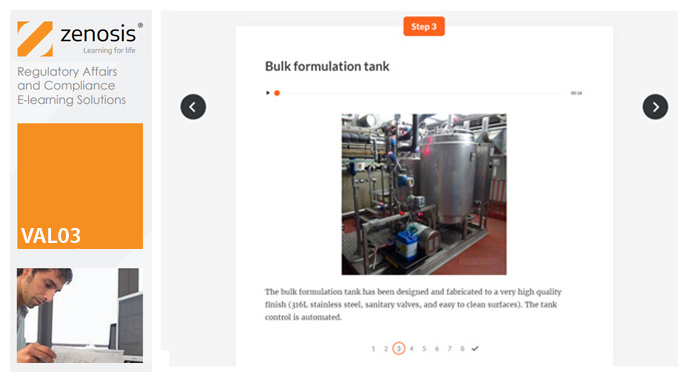
VAL03: Commissioning and Installation Qualification
By Zenosis
Before equipment can be used routinely in production, it must first be commissioned and, if necessary, undergo Installation Qualification (IQ). This module describes commissioning and IQ requirements and procedures in the medicines and healthcare products industries. It follows the activities of a typical validation team as they carry out a project for a pharmaceutical company.
SUB15: The Biologics License Application (BLA) for Marketing Approval in the USA
By Zenosis
This module describes the requirements that must be met to obtain licensure of a biological product. Subjects covered include the regulatory context, the content and format of the BLA submission, the review process, and provisions for expedited development and review.

SUB14: The Regulatory Pathway to Licensure of Follow-on Biologics (Biosimilars) in the USA
By Zenosis
The regulation of biological medicinal products is governed by different laws from those that apply to small-molecule synthetic drugs. Producing faithful copies of therapeutic proteins is more challenging than producing generic drugs. The US legal framework for the licensure of follow-on biologics, and accompanying regulatory guidance from the Food and Drug Administration (FDA), have been established only in recent years.

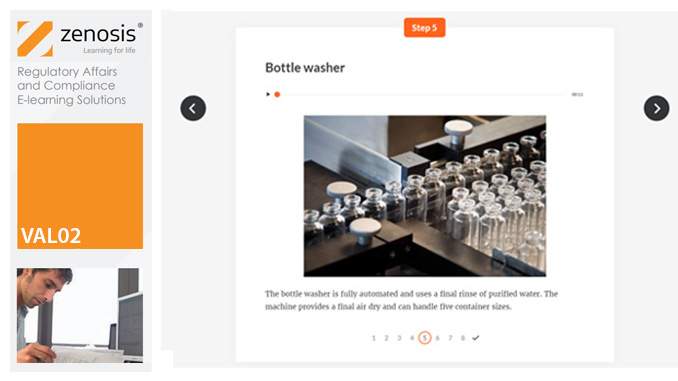
VAL02: Validation Plans and Documentation
By Zenosis
Essential to validation is the provision of documented evidence verifying that manufacturing processes will consistently result in products meeting predetermined quality standards. This module describes the purpose, content and use of validation master plans, project validation plans, and other documentation for validation projects in the medicines and healthcare products industries. It describes the activities of a typical validation team as they carry out a project for a pharmaceutical company.
VAL07: Computer Systems Validation, Part 2: Implementation
By Zenosis
This module describes the design, development and installation phase, the validation phase, and the operation and maintenance phase of the validation of computerised systems in medicines and healthcare products manufacturing environments. It continues to follow the progress of a pharmaceutical company's project to validate a new dispensary control system.
VAL06: Computer Systems Validation, Part 1: Planning
By Zenosis
In the medicines and healthcare products industries, computerised systems used in automated manufacturing or laboratory processes to which Good Manufacturing Practice requirements apply need to be validated. This module describes the planning of such validation. It follows the work of a pharmaceutical company's team as they validate the dispensary control system for a new production line.

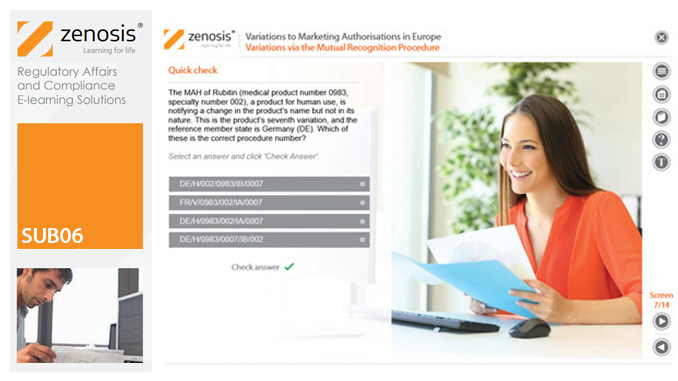
SUB06: Variations to Marketing Authorisations in Europe
By Zenosis
Changes to the terms of marketing authorisations for medicinal products, called variations in Europe, must be notified to or approved by the relevant regulatory authorities. Variations include changes to the composition of products, their manufacturing processes, the way they are used, or the indications for which they are authorised. Common approaches are adopted within the European Economic Area to variations to marketing authorisations approved through the Centralised, Decentralised or Mutual Recognition Procedures. Recent legislation has substantially modified the regulatory requirements and extended them to purely national authorisations by member states. This module, which is fully up to date with the new legislation, covers the classification of variations into their several types and the regulatory requirements, guidance and procedures to be followed for each type.
ESS01: Essentials of EU and US Regulatory Affairs for Human Medicinal Products
By Zenosis
This foundation-level module is the ideal introduction for new entrants to the field of pharmaceutical regulatory affairs and compliance. It describes the principal requirements that must be satisfied to gain and maintain approval to market medicinal products in the USA and Europe. The legal framework and the roles of major players in regulation are presented. The life-cycle of a drug is outlined. The various procedures available for assessment and approval of products are described and their requirements outlined. Obligations to be fulfilled after marketing approval are discussed.

SAM02: Regulatory Requirements and Guidance on Advertising and Promotion of Prescription Drugs in the USA
By Zenosis
In this course we explain how to advertise and promote prescription drugs in various media, whether to healthcare professionals or consumers, in compliance with legal requirements and guidance from the FDA.

SAM04: Marketing of Prescription Drugs in the USA – Interactions with Healthcare Professionals
By Zenosis
The heaviest legal penalties imposed on drug companies concern interactions with healthcare professionals in the context of prescription drug marketing, notably for violations of the Anti-Kickback Statute and the False Claims Act. Monetary penalties have amounted to billions of dollars in some cases.
