- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
2185 Professional Development courses delivered On Demand
Financial Analyst - QLS Endorsed Level 4 Diploma
By Imperial Academy
Level 4 Diploma(FREE QLS Endorsed Certificate)| 11 CPD Courses+11 PDF Certificates| 140 CPD Points|CPD & CiQ Accredited

Social Worker Training
By Imperial Academy
Level 5 Diploma(FREE QLS Endorsed Certificate)| 12 CPD Courses+12 PDF Certificates| 150 CPD Points| CPD & CiQ Accredited


Level 3 Award in Education and Training City and Guilds 6502 (Previously PTTLS)
By Harpar Qualifications Ltd
This course is suitable if you work or want to work as a teacher/trainer/tutor in Further Education Colleges, adult and community education, the voluntary sector, commerce, industry, the public sector or HM forces.


VAL01: Introduction to Validation
By Zenosis
Validation of equipment, services, systems and processes is vitally important in the medicines and healthcare products industries. Regulatory authorities require documented evidence that manufacturing processes will consistently result in products meeting predetermined quality standards. This module provides an introduction to validation and to the regulations and guidance that apply to it. It describes the activities of a typical validation team as they carry out a project for a pharmaceutical company.
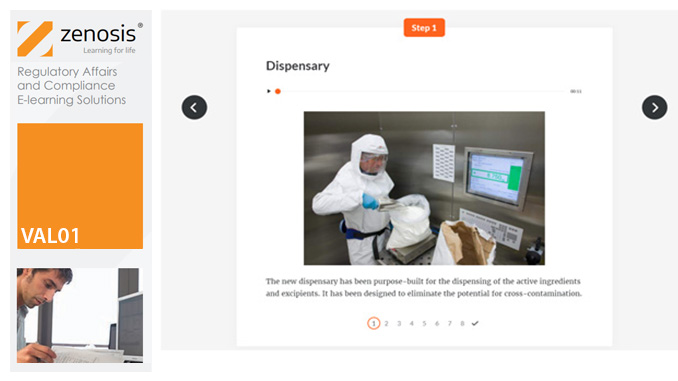
VAL03: Commissioning and Installation Qualification
By Zenosis
Before equipment can be used routinely in production, it must first be commissioned and, if necessary, undergo Installation Qualification (IQ). This module describes commissioning and IQ requirements and procedures in the medicines and healthcare products industries. It follows the activities of a typical validation team as they carry out a project for a pharmaceutical company.
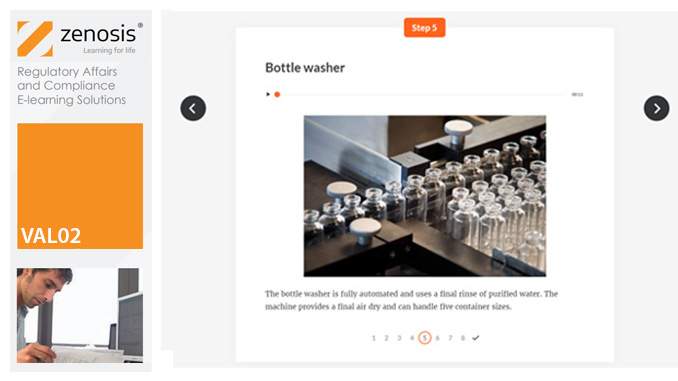
VAL02: Validation Plans and Documentation
By Zenosis
Essential to validation is the provision of documented evidence verifying that manufacturing processes will consistently result in products meeting predetermined quality standards. This module describes the purpose, content and use of validation master plans, project validation plans, and other documentation for validation projects in the medicines and healthcare products industries. It describes the activities of a typical validation team as they carry out a project for a pharmaceutical company.
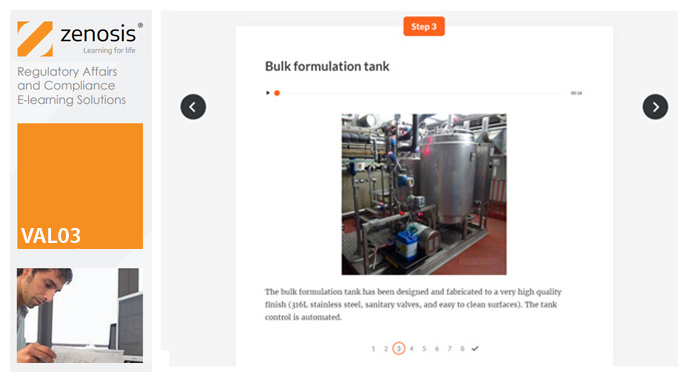
VAL05: Equipment Cleaning Validation
By Zenosis
Manufacturers of medicines and healthcare products must establish, validate and maintain an equipment cleaning programme. This is a regulatory requirement because validated cleaning procedures contribute to the assurance of product purity and safety. This module provides a comprehensive account of equipment cleaning validation requirements and procedures. It follows the work of a pharmaceutical company's validation team as they establish and validate the cleaning program for a new production line.
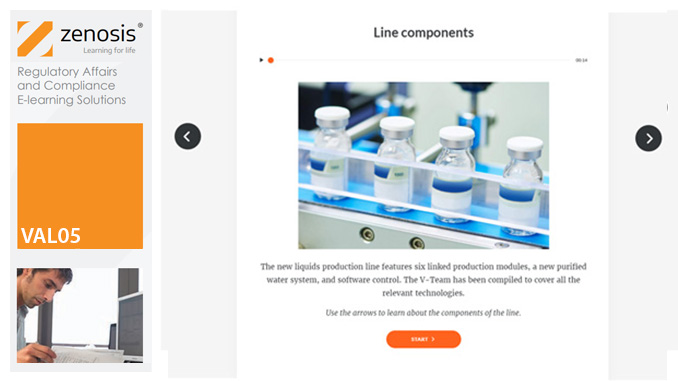
SUB15: The Biologics License Application (BLA) for Marketing Approval in the USA
By Zenosis
This module describes the requirements that must be met to obtain licensure of a biological product. Subjects covered include the regulatory context, the content and format of the BLA submission, the review process, and provisions for expedited development and review.
