- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
288 Pharmacology courses
Epilepsy and Buccal Midazolam
By Wise Campus
Epilepsy and Buccal Midazolam Are you interested in learning about buccal midazolam for managing acute seizures in epilepsy patients? Then our epilepsy and buccal midazolam course will be your perfect guide. The principles of epilepsy, identifying various seizure types, and the pharmacology of midazolam are all covered in this course on epilepsy and buccal midazolam. This course on epilepsy and buccal midazolam also covers dose calculations and appropriate delivery methods. Additionally, safety procedures for buccal midazolam are explained in the Epilepsy and Buccal Midazolam course. This epilepsy and buccal midazolam course also explains documentation procedures, legal implications, and possible side effects. Without further delay, participate in our epilepsy and buccal midazolam course and enhance your knowledge in this field! Learning outcome of epilepsy and buccal midazolam This epilepsy and buccal midazolam course teaches: Comprehensive training on epilepsy and buccal midazolam. Handling epilepsy, emergency response, and first aid are included in this epilepsy and buccal midazolam course. This epilepsy and buccal midazolam course also explains the administration of medication. How to provide support and resources for this disorder is elaborated on in this epilepsy and buccal midazolam course. Throughout this epilepsy and buccal midazolam course, you can learn safety and lifestyle management techniques. Legal and ethical considerations are part of this epilepsy and buccal midazolam course. This epilepsy and buccal midazolam course provides simulation and case studies for practical knowledge of this field. Special Offers of this Epilepsy: Epilepsy Course This Epilepsy: Epilepsy Course includes a FREE PDF Certificate. Lifetime access to this Epilepsy: Epilepsy Course Instant access to this Epilepsy: Epilepsy Course Get FREE Tutor Support to this Epilepsy: Epilepsy Course Epilepsy and Buccal Midazolam Unlock your potential with our Epilepsy: Epilepsy and Buccal Midazolam course! Gain an in-depth understanding of Epilepsy: Epilepsy principles and learn to identify various seizure types with precision. This Epilepsy: Epilepsy course delves into the pharmacology of midazolam, ensuring accurate dose calculations and effective delivery methods. Master safety procedures for buccal midazolam in our comprehensive Epilepsy: Epilepsy training. Additionally, explore essential documentation procedures, legal implications, and potential side effects in this Epilepsy: Epilepsy course. Enroll now to enhance your expertise and provide exceptional care in the field of Epilepsy: Epilepsy management! Who is this course for? Epilepsy and Buccal Midazolam This epilepsy: epilepsy and buccal midazolam training is intended for those who work in the field of epilepsy: epilepsy care and may need to provide buccal midazolam to patients who are having seizures. Requirements Epilepsy and Buccal Midazolam To enrol in this Epilepsy: Epilepsy Course, students must fulfil the following requirements. To join in our Epilepsy: Epilepsy Course, you must have a strong command of the English language. To successfully complete our Epilepsy: Epilepsy Course, you must be vivacious and self driven. To complete our Epilepsy: Epilepsy Course, you must have a basic understanding of computers. A minimum age limit of 15 is required to enrol in this Epilepsy: Epilepsy Course. Career path Epilepsy and Buccal Midazolam With the epilepsy: epilepsy and buccal midazolam course qualification, you can work as a clinician educator, care coordinator, or epilepsy expert, with chances for research, leadership, and policy.

GLP02 - Good Quality Control Laboratory Practice
By Zenosis
The medicinal products industry is heavily regulated by governments. Within the industry’s Good Manufacturing Practice (GMP) framework, analytical laboratories engaged in quality control (QC) of starting materials, intermediates, bulk products, finished products, and packaging need to comply with relevant GMP standards. We refer to these as Good Quality Control Laboratory Practice, or GQCLP. Regulatory authorities inspect laboratories to confirm that they meet the standards. This course explains how to comply with GQCLP, and it provides advice on laboratory work in general.

GLP01 - Good Laboratory Practice
By Zenosis
The purpose of GLP is to provide assurance of the quality and reliability of nonclinical study data. GLP covers the planning, performance, monitoring, recording and reporting of studies. Regulatory authorities typically require GLP rules to be followed for nonclinical studies intended to support an application for approval of clinical research or marketing of a product containing the test item. This course outlines the history of GLP and explains why it is important, identifies the penalties that may be incurred for noncompliance, and sets out requirements that need to be met. Learners are also referred to the two main sources of GLP rules: The Organisation for Economic Co-operation and Development’s Principles on Good Laboratory Practice and US Regulation 21 CFR 58: Good Laboratory Practice for Nonclinical Laboratory Studies.

PV04: Signal Detection and Management in Pharmacovigilance
By Zenosis
This module provides a guide to signal detection and management for approved products. The subject is presented as a process comprising four stages: signal detection, signal validation, signal analysis and prioritisation, and risk assessment and minimisation.

SUB04: Preparing Submissions in the Common Technical Document (CTD) Format
By Zenosis
The CTD is the internationally recognised standard format for submissions to medicines regulatory authorities. In the European Economic Area, the USA and Canada, the CTD, in its electronic format (eCTD), is mandatory for all applications for marketing approval and all subsequent related submissions. The CTD is accepted in many other countries, being mandatory for new prescription medicines in some. This module explains the rationale for the CTD and provides guidance on its structure and format and the ways in which it is used.

GMP01: An Introduction to Good Manufacturing Practice for Medicinal Products
By Zenosis
Good Manufacturing Practice (GMP) is a set of rules for medicines manufacturers to follow so that their products are safe, effective, and of good quality. The rules may be written into law or set out in guidance documents from regulatory authorities. Regulators will not allow medicinal products to be placed, or to remain, on the market in their country unless the products can be shown to be manufactured in compliance with GMP. To this end, they carry out inspections of manufacturing plants. Companies that persistently commit serious breaches of GMP requirements have suffered huge fines.
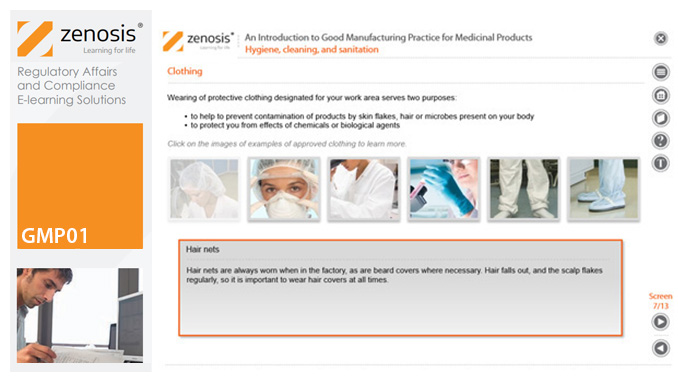
GMP04: Good Manufacturing Practice for the Warehouse
By Zenosis
The warehouse plays a crucial role in a medicinal products factory. This module explains the requirements of Good Manufacturing Practice (GMP) for the warehouse, and how to comply with them.

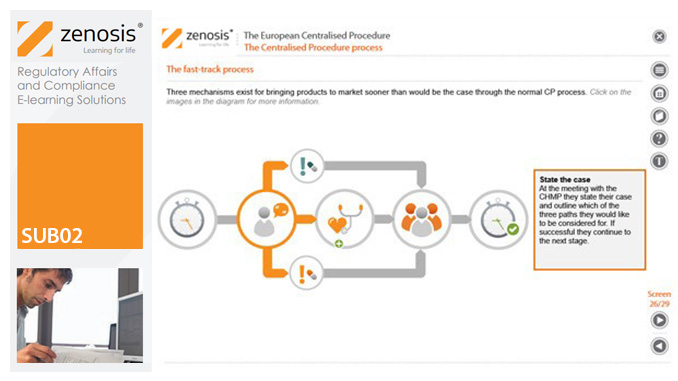
SUB02: The European Centralised Procedure (CP)
By Zenosis
The Centralised Procedure (CP) is one of three routes available to applicants to gain multinational marketing authorisation within the European Economic Area (EEA) on the basis of a single application. In the CP, one successful application leads to a marketing authorisation being issued by the European Commission that applies throughout the EEA. The CP is mandatory for certain types of products.
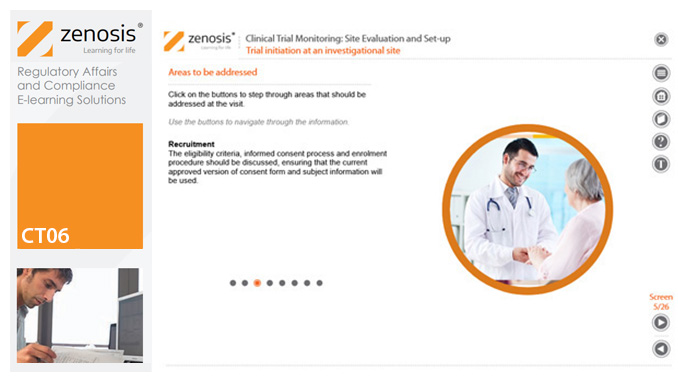
CT06: Clinical Trial Monitoring: Site Evaluation and Setup
By Zenosis
The sponsor of a clinical trial needs to reach agreement with clinical investigators to conduct the trial. The suitability of investigators and their institutional sites, typically hospitals, has to be evaluated, and the trial has to be set up at each site. This module describes the processes involved, focusing particularly on the role of a Clinical Research Associate (CRA) employed or contracted by the sponsor to monitor the trial.
PKPD01: An Introduction to Pharmacokinetics and Pharmacodynamics in Drug Development and Registration
By Zenosis
Pharmacokinetic (PK) and pharmacodynamic (PD) studies provide a bridge between science and medicine in the development of a drug. In this module we describe the role of in-vivo PK and PD studies in a drug development programme, set out the uses to which the findings can be put, and discuss their implications for clinical development and application for marketing approval.
