- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
Courses in Cardiff
We couldn't find any listings for your search. Explore our online options below.
Know someone teaching this? Help them become an Educator on Cademy.
Online Options
Show all 42Data Protection for Clinical Trials and Medical Research
By Computer Law Training
Data Protection and Clinical Trials

CT12: How to Conduct Clinical Research Under the EU Clinical Trials Regulation
By Zenosis
This course describes the requirements that must be met by, and options available to, the sponsor during the conduct of an authorised clinical trial. It identifies the various interactions with MSCs that occur via the Clinical Trials Information System (CTIS), and it summarises and links to the extensive guidance available from the European Commission and the European Medicines Agency. Its companion course CT11 sets out the European legal and regulatory context for clinical trials and describes how to apply via the CTIS for authorisation to conduct trials. The two courses therefore provide an ideal foundation for understanding and complying with the new law.

CT11: How to Gain Authorisation for Clinical Research Under the EU Clinical Trials Regulation
By Zenosis
This course sets out the procedures that sponsors need to follow to gain authorisation to conduct clinical trials under the Regulation, and it summarises and links to the extensive guidance available from the European Commission and the European Medicines Agency. Its companion course CT12 sets out the procedures that sponsors need to follow to conduct authorised clinical trials in compliance with the Regulation. The two courses therefore provide an ideal foundation for understanding and complying with the new law.
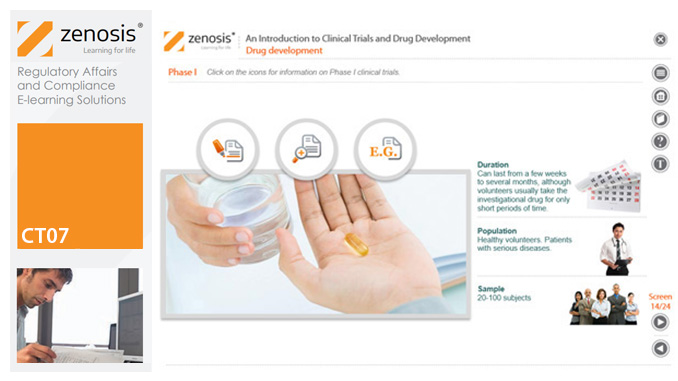
CT07: An Introduction to Clinical Trials and Drug Development
By Zenosis
This module provides an understanding of how clinical trials fit into the drug development process. It outlines the key historical events leading to the development of controlled clinical trials. It specifies the purpose of trials, outlines their features, and identifies codes and regulations that apply to them. Finally, it describes the environment of cost control in which the modern pharmaceutical industry operates.
CT04a - Clinical trials in drug development
By Zenosis
New drug development requires major investment in capital, human resources and technical expertise. Strict adherence to regulations on testing and manufacturing standards is also required before a new drug can be marketed. One of the greatest challenges in conducting clinical trials is that of efficiency. As trials become more comprehensive, involving large numbers of participants globally, their duration is prolonged and costs increase. The longer trials last, the shorter is the patent life remaining after market approval and the longer patients must wait for the new product. This short course covers the key components of clinical trials and how these requirements interact with the drug development cycle.

CT01: How to Gain and Maintain Approval for Clinical Research Under the EU Clinical Trials Directive
By Zenosis
To conduct a clinical trial in the European Economic Area under the Clinical Trials Directive the sponsor must apply for authorisation from the national competent authority (i.e. medicines regulator), and favourable opinion must be obtained from a research ethics committee, in each member state in which the trial is to take place. This module sets out the requirements for successful compilation, submission and maintenance of the applications.
CT13: Safety Reporting in Clinical Trials
By Zenosis
This course explains the regulatory requirements for the reporting of adverse events and suspected adverse reactions in clinical trials. It describes how investigators should report to sponsors, and how sponsors should report to regulatory authorities and other stakeholders in the safety of investigational products. It explains how events are characterized as serious or non-serious, expected or unexpected, and it distinguishes the requirements for each category. It describes controlled vocabularies used for coding of events in reports.
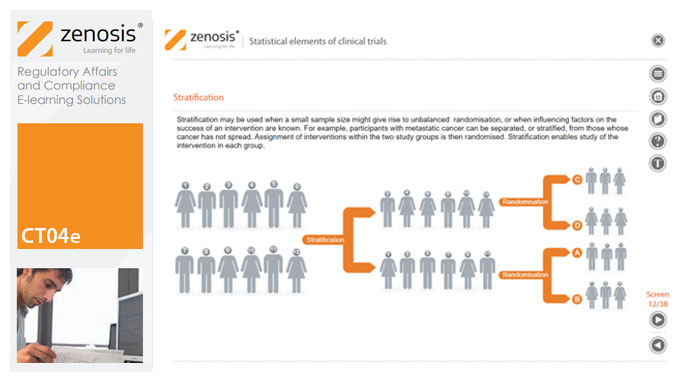
CT04e - Statistical elements of clinical trials
By Zenosis
Analytical statistical elements are essential concepts in the design of clinical trials. This analysis helps us to understand whether a conclusion from a study of a sample of the target population applies generally to that population as a whole. In particular, it helps us to answer the question: Did the treatment effect in the given study occur just by chance? The statistical elements of a well-controlled study minimise the chances of drawing the wrong conclusions, by providing clear thresholds for such errors. The basic statistical elements of a clinical trial include eligibility criteria, randomisation, sample size, power, and blinding, and these are discussed in this short course.
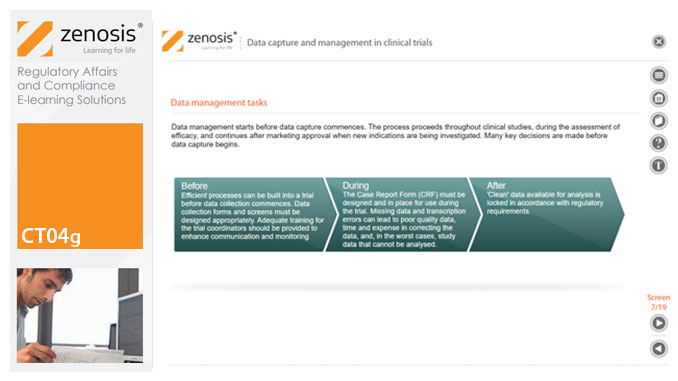
CT04g - Data capture and management in clinical trials
By Zenosis
Capture and management of clinical trial data is a challenge. The industry is under pressure to obtain and analyse such data more quickly, while maintaining data integrity, so that products can be brought to market sooner. Effective planning and adequate resources can ensure clinical trials yield high quality data within strict timelines and budget requirements, at the same time satisfying regulatory standards. This short course describes the purpose of data capture and explores efficiencies in data management as part of the evolving regulatory landscape.
CT14: Clinical Trial Safety Reporting Requirements in the EU and USA
By Zenosis
This course sets out the legal and regulatory requirements for safety reporting in clinical trials of medicinal products under the jurisdictions of the European Union and the USA. It builds on the foundation laid by our companion course CT13, Safety Reporting in Clinical Trials, and provides greater detail of specific requirements in those jurisdictions.
