- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
128 Good Manufacturing Practice (GMP) courses in Cardiff delivered Online
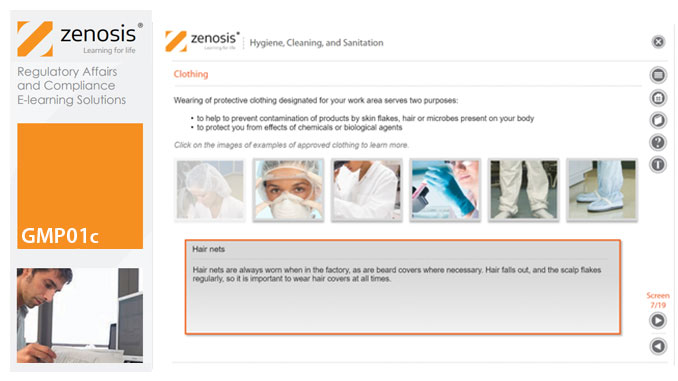
GMP01c - Hygiene, cleaning, and sanitation
By Zenosis
Prevention of contamination is one of the most important goals of GMP. Contamination of product is often difficult to detect, so GMP rules emphasise preventive measures, including: attention to personal health and hygiene, and the wearing of special clothing, by staff; and cleaning and sanitation of premises and equipment. In this short course we set out the basics of GMP requirements in these vital areas.
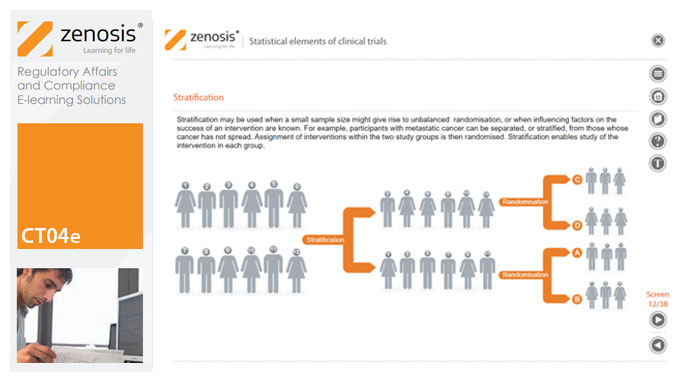
CT04e - Statistical elements of clinical trials
By Zenosis
Analytical statistical elements are essential concepts in the design of clinical trials. This analysis helps us to understand whether a conclusion from a study of a sample of the target population applies generally to that population as a whole. In particular, it helps us to answer the question: Did the treatment effect in the given study occur just by chance? The statistical elements of a well-controlled study minimise the chances of drawing the wrong conclusions, by providing clear thresholds for such errors. The basic statistical elements of a clinical trial include eligibility criteria, randomisation, sample size, power, and blinding, and these are discussed in this short course.
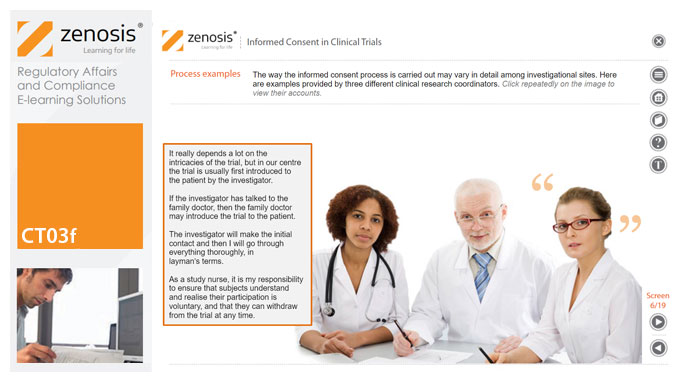
CT03f - Informed consent in clinical trials
By Zenosis
Informed consent in clinical research is an ethical and regulatory requirement. A research subject must enter a study voluntarily, be informed about risks and benefits, and understand the difference between investigation and treatment. Subjects must not be coerced into enrolment, nor must they be enticed by exaggerated claims of benefit. Before they can enrol, all potential subjects must agree, in writing, to participate. In addition to ethical and regulatory imperatives, the potential for litigation by subjects further highlights the importance of rigorous adherence to informed consent principles. In this short course we set out the principles and requirements and provide examples of practical issues confronting healthcare professionals and subjects.
CT03d - Clinical trial sponsor’s GCP responsibilities
By Zenosis
The sponsor of a clinical trial takes responsibility for its initiation, management, and/or financing. A sponsor may transfer any or all of the sponsor’s trial-related duties and functions to a contract research organisation, but the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor. Duties and functions discussed in this short course include trial design, selection of investigators, QA and QC, data handling and record keeping, finance and compensation, regulatory submissions, management of investigational product(s), safety reporting, monitoring, audit, dealing with noncompliance, and clinical trial reports. ICH guideline E6 (revision 2) encourages sponsors to adopt a risk-based approach to managing the quality of trials. We discuss this approach in general, and aspects such as risk-based monitoring in particular.
CT03c - Clinical trial documentation
By Zenosis
Regulatory authorities tend to abide by the maxim that ‘If it isn’t documented, it didn’t happen’. Rigorous documentation of all aspects of a clinical trial is necessary to provide evidence of GCP and compliance with regulatory requirements, as well as enabling effective management of the trial. In this short course we describe important examples of the documents designated by ICH GCP as essential to the conduct of a clinical trial.
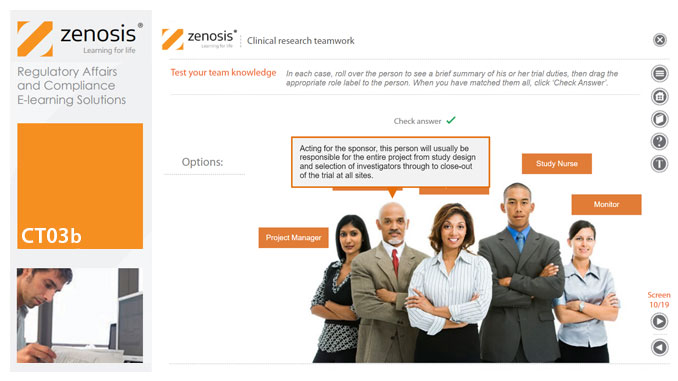
CT03b - Clinical research teamwork
By Zenosis
A clinical trial, particularly a late-phase commercial study, is a major project requiring collaboration between the sponsor and staff or contractor, on the one hand, and the clinical investigator(s) and other healthcare professionals on the other. Good communication among all parties is essential. In this short course we introduce the major roles in a typical clinical research project and outline their duties.
GMP01d - Documentation
By Zenosis
Comprehensive documentation of procedures, formulas, work instructions, and specifications, and thorough recording of batch data, are fundamental requirements of GMP. In this short course we explain why documentation is so important, identify different types of document required, and set out some simple rules for recording and correcting data.
Good Clinical Practices: A Practical Guide to GCP Compliance
By Xpert Learning
About Course Understand the Ethical and Regulatory Framework for Conducting Clinical Trials Course Description This comprehensive Good Clinical Practices (GCP) course provides a thorough understanding of the ethical and regulatory principles governing clinical trials. It delves into the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines, ensuring you grasp the essential standards for conducting clinical research. Course Objectives By the end of this course, you will be able to: Articulate the definition, purpose, and historical context of GCP. Explain the importance of GCP in protecting human subjects and ensuring data integrity. Identify key international organizations involved in establishing GCP standards. Apply ethical principles and informed consent procedures in clinical research. Understand the ICH E6(R2) guidelines on Integrated Addendum to Good Clinical Practice. Design and conduct clinical trials according to ICH E8 and E9 guidelines. Manage and report clinical trial data in compliance with ICH E6(R2) guidelines. Implement safety monitoring and adverse event reporting procedures as per ICH E6(R2) and E6(R3) guidelines. Navigate regulatory compliance and inspections related to clinical trials. Target Audience This course is designed for individuals who: Are interested in pursuing a career in clinical research or clinical trial management. Work in the pharmaceutical, biotechnology, or medical device industry. Seek to gain a comprehensive understanding of GCP principles and practices. Prerequisites No prior experience in clinical research is required. However, a basic understanding of research methodology and medical terminology is recommended. Please Note: This course is strictly theoretical and does not qualify participants for clinical practice in the industry. Additional training and certifications may be required for hands-on experience. What Will You Learn? Articulate the definition, purpose, and historical context of GCP. Explain the importance of GCP in protecting human subjects and ensuring data integrity. Identify key international organizations involved in establishing GCP standards. Apply ethical principles and informed consent procedures in clinical research. Understand the ICH E6(R2) guidelines on Integrated Addendum to Good Clinical Practice. Design and conduct clinical trials according to ICH E8 and E9 guidelines. Manage and report clinical trial data in compliance with ICH E6(R2) guidelines. Implement safety monitoring and adverse event reporting procedures as per ICH E6(R2) and E6(R3) guidelines. Navigate regulatory compliance and inspections related to clinical trials. Course Content Introduction to Good Clinical Practice (GCP) and ICH Guidelines Introduction to Good Clinical Practice (GCP) and ICH Guidelines Ethical Principles, Informed Consent, and ICH E6(R2) Ethical Principles, Informed Consent, and ICH E6(R2) Designing and Conducting Clinical Trials with ICH E8 and E9 Designing and Conducting Clinical Trials with ICH E8 and E9 Data Management and Reporting with ICH E6(R2) Data Management and Reporting with ICH E6(R2) Safety and Monitoring in Clinical Trials with ICH E6(R2) and E6(R3) Safety and Monitoring in Clinical Trials with ICH E6(R2) and E6(R3) Regulatory Compliance, Inspections, and ICH E6(R3) Regulatory Compliance, Inspections, and ICH E6(R3) A course by Xpert Learning RequirementsA basic understanding of research methodology and medical terminology is recommended. Audience Individuals who are interested in pursuing a career in clinical research or clinical trial management. Individuals who work in the pharmaceutical, biotechnology, or medical device industry. Individuals who seek to gain a comprehensive understanding of GCP principles and practices. Audience Individuals who are interested in pursuing a career in clinical research or clinical trial management. Individuals who work in the pharmaceutical, biotechnology, or medical device industry. Individuals who seek to gain a comprehensive understanding of GCP principles and practices.

