- Professional Development
- Medicine & Nursing
- Arts & Crafts
- Health & Wellbeing
- Personal Development
36981 Courses
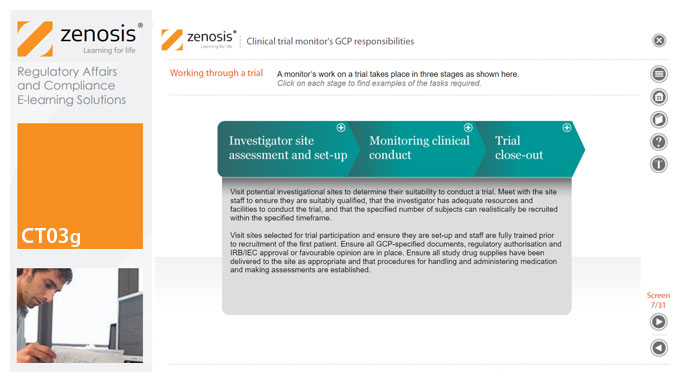
CT03g - Clinical trial monitor’s GCP responsibilities
By Zenosis
A clinical trial monitor acts on behalf of the sponsor to support investigational site personnel, verify the accuracy of data recorded, and ensure that the trial is conducted in compliance with the protocol, GCP and other study specific requirements. He or she acts as the ‘eyes and ears’ of the sponsor at the investigational site and provides the main channel of communication between sponsor and investigator. This short course explores the responsibilities of the monitor and provides insight into key challenges. We discuss assessment of investigators and investigational sites, education and trial initiation, monitoring of clinical conduct, including CRF review and source document verification, and trial close-out. We discuss noncompliance and how to deal with it.
CT04a - Clinical trials in drug development
By Zenosis
New drug development requires major investment in capital, human resources and technical expertise. Strict adherence to regulations on testing and manufacturing standards is also required before a new drug can be marketed. One of the greatest challenges in conducting clinical trials is that of efficiency. As trials become more comprehensive, involving large numbers of participants globally, their duration is prolonged and costs increase. The longer trials last, the shorter is the patent life remaining after market approval and the longer patients must wait for the new product. This short course covers the key components of clinical trials and how these requirements interact with the drug development cycle.

CT03a - ICH, harmonisation, and principles of Good Clinical Practice
By Zenosis
Good Clinical Practice (GCP) is a set of internationally recognised ethical and scientific quality requirements for designing, conducting, recording and reporting clinical trials. Compliance with GCP principles is required by regulatory authorities in many countries for the authorisation of clinical trials and the acceptance of their data in applications for marketing approval. The International Council for Harmonisation's guideline E6, often referred to as ICH GCP, is the international standard specification for Good Clinical Practice. In this short course we describe the ICH’s role in the harmonisation of regulations, introduce its guideline E6, and set out the principles of GCP.
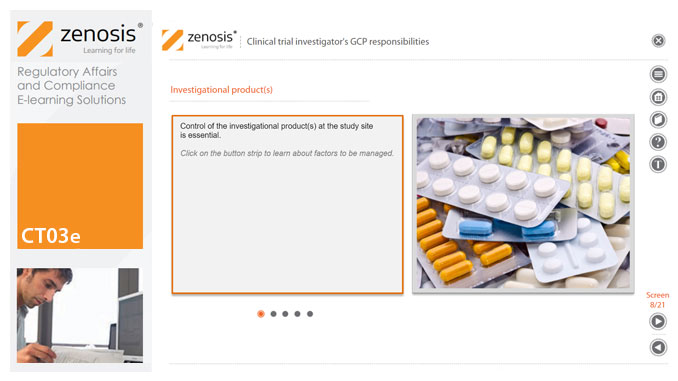
CT03e - Clinical trial investigator’s GCP responsibilities
By Zenosis
A clinical investigator is responsible for conducting the clinical trial in compliance with the study protocol, GCP, medical ethics, and applicable legal requirements. The clinical research community expects that investigators and clinical staff are fully trained in GCP. Duties and functions discussed in this short course include: provision of adequate resources; liaison with IRB/IEC; compliance with protocol; management of investigational product(s), informed consent and data records; and safety reporting.
GMP01a - GMP – what and why
By Zenosis
Good Manufacturing Practice (GMP) is a set of rules for medicines manufacturers to follow so that their products are safe, effective, and of good quality. Everyone who works in a processing, quality control, packaging, or warehouse environment for a pharmaceutical or biotechnology company, or one of their contractors, must understand why GMP is important, how it applies to them, and how to comply with it. This short course explains what GMP is and why it is important, and it gives some lessons from history. It introduces the regulations and guidance documents that are the source of GMP rules. Finally, it touches on regulatory inspections and the consequences that can arise from failure to comply with GMP requirements.

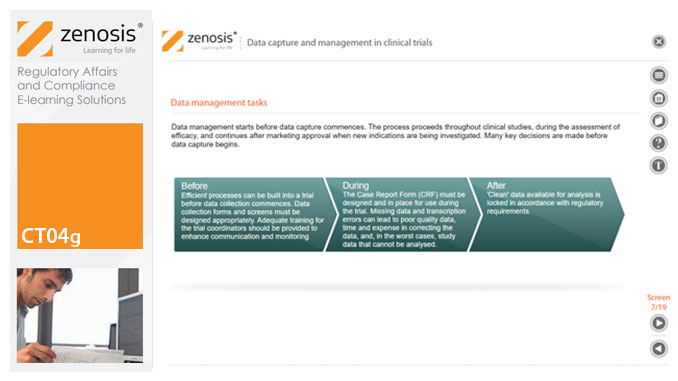
CT04g - Data capture and management in clinical trials
By Zenosis
Capture and management of clinical trial data is a challenge. The industry is under pressure to obtain and analyse such data more quickly, while maintaining data integrity, so that products can be brought to market sooner. Effective planning and adequate resources can ensure clinical trials yield high quality data within strict timelines and budget requirements, at the same time satisfying regulatory standards. This short course describes the purpose of data capture and explores efficiencies in data management as part of the evolving regulatory landscape.
CT04c - Clinical trial preparation
By Zenosis
The demands on quality from clinical trials are increasing. Quantitative aspects of clinical trials, such as the mass of study data to be collected, the multiple investigational sites, and the need to meet predetermined timelines, often supersede qualitative features. Therefore, addressing basic requirements for quality management is essential when preparing a clinical trial. This short course describes the core elements required for the establishment of a clinical trial and provides an overview of the role of the sponsor in supporting and improving trial quality.
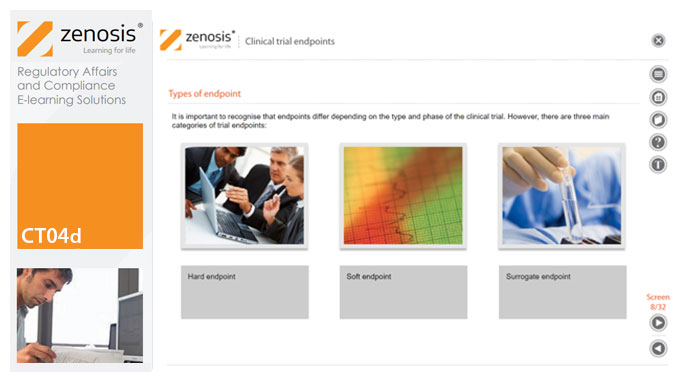
CT04d - Clinical trial endpoints
By Zenosis
In clinical trials, endpoints are measurements to evaluate the results of a new treatment, at an individual patient level. The study data can be extrapolated to patient populations on the basis of clinical similarities to patients participating in the trial. When clinical trial data have been obtained, focus is on the trial endpoints; more specifically, the focus is on whether the trial met or failed the primary endpoint specified before the trial started. The purpose and various types of endpoints are discussed in this short course.
CT04b - Clinical protocol design
By Zenosis
Clinical trial protocols are an essential part of clinical trial design. Protocol documents are critical to conducting safe and cost-effective investigations. Protocol documents are large and complex, containing comprehensive information relating to purpose, design and conduct of a clinical trial. Aspects of a protocol include patient eligibility criteria, and treatment specifications. This short course provides an overview of clinical trial protocols. Opportunities to improve a clinical trial protocol for regulatory approval are also discussed.
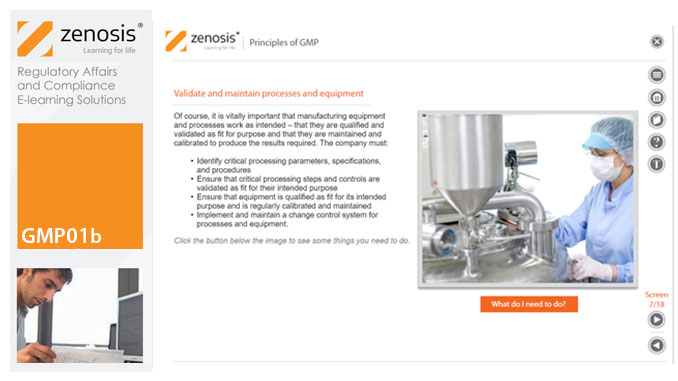
GMP01b - Principles of GMP
By Zenosis
In this short course we present an overview of the main principles of GMP, and we outline some things that manufacturing personnel need to do to comply with requirements. We identify the principal goals of GMP as: prevention of contamination; prevention of mix-ups; scrupulous documentation; validation and maintenance of processes and equipment; quality assurance by an independent unit; and training. We place GMP in the context of a company’s quality management system.